The benefits of coupling physics to robotics, quantum chemistry & mathematics
November 21, 2024
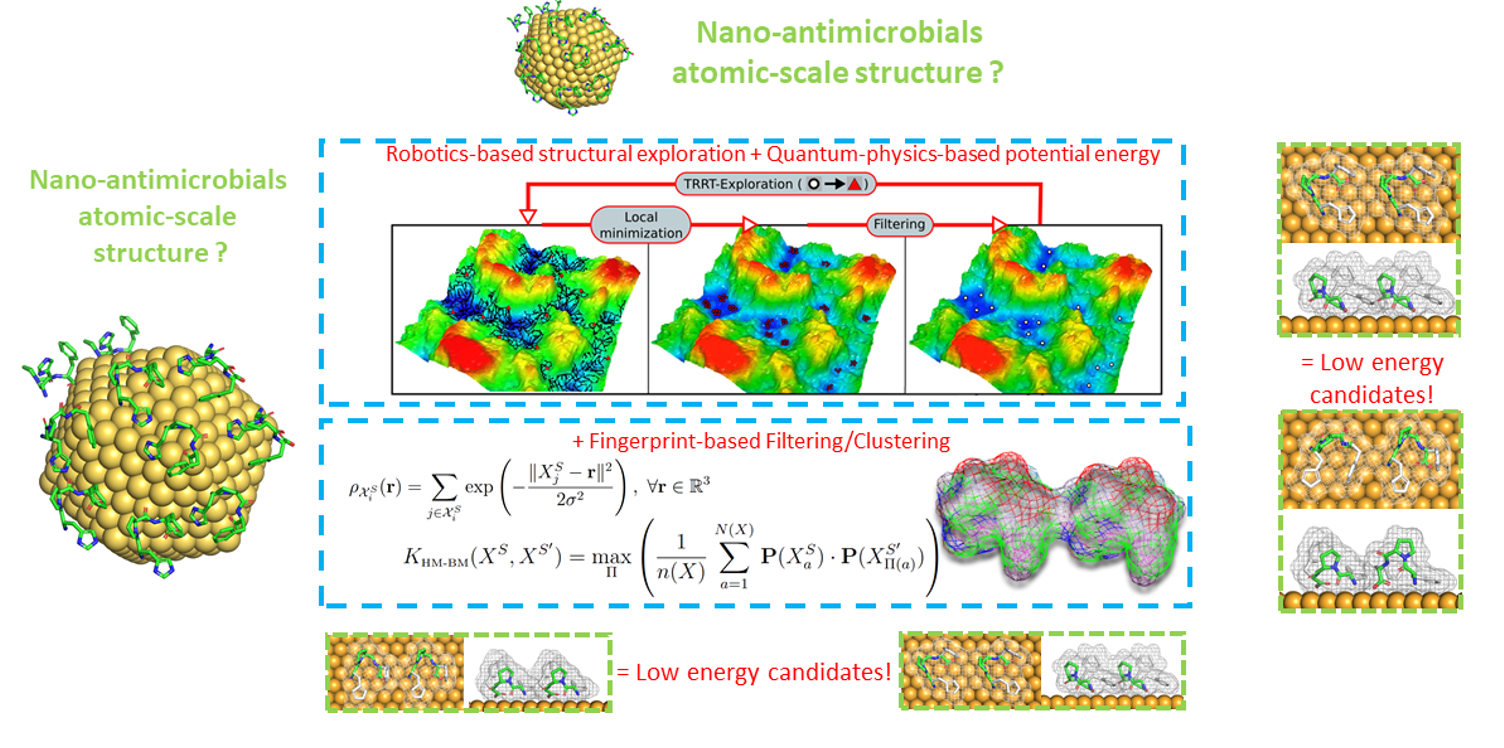
By adapting and combining robotics-inspired structural exploration algorithms, energy potentials from quantum chemistry and mathematical tools to assess the (dis)similarity between two complex hybrid nano-systems, we are gaining understanding of their structural and electronic properties, paving the way for the rational design of nano-antimicrobials, highly promising objects for combating resistant microorganisms.
The development of nano-antimicrobials (metal nanoparticles functionalized with bioactive molecules) is a promising strategy in the international fight against resistant microorganisms. However, their atomic and electronic structures are poorly understood, which is a major obstacle to their rational design.
In order to achieve a structural understanding of functionalized surfaces, and to be able to apprehend the electronic mechanisms at play, we have adapted/optimized methods to explore their conformations in an efficient and non-redundant way, and interfaced it with a quantum chemistry code. The robotics-inspired algorithm, named IGLOO (Iterative Global Exploration and Local Optimization), uses tree-based exploration and dynamically adjusts its exploration strategy to scan the energy landscape evenly and focus on promising regions, avoiding redundant exploration. The interfaced quantum chemistry code, deMonNano, enables the energy calculations required for IGLOO exploration to be performed with a DFTB quantum energy potential.
Within such structural explorations, a major challenge is to assess the (dis)similarity of the nano-systems explored to obtain a clustering of the latter. Since the nano-antimicrobials we model have the particularity of being homomolecular (i.e. containing a large number of a given molecule), we have adapted a fingerprint-based method called SOAP (Smooth Overlap of Atomic Positions), taking advantage of the structural architecture of homomolecular systems to minimize irrelevant comparisons of atomic environments. This has enabled us to reduce computational costs and noise in (dis)similarity measurements, with the consequence of producing more meaningful clustering results than native methods, by better capturing key structural differences between states.
Our next steps will be to carry out the developments needed to perform these explorations not only on flat surfaces, but also on nanoparticles functionalized by a large number of ligands.
This work is being carried out as part of a collaboration between LAAS, LCPQ, IMT, MPI-Stuttgart and CEMES.
Contact:
Nathalie Tarrat | nathalie.tarrat[at]cemes.fr
Publications:
Exploring molecular energy landscapes by coupling DFTB potential with a tree-based stochastic algorithm: Investigation of the conformational diversity of Phthalates
V. Milia, N. Tarrat, C. Zanon, J. Cortés, and M. Rapacioli
Journal of Chemical Information and Modelling 64 (2024) 3290
DOI: https://doi.org/10.1021/acs.jcim.3c01981
Dependence of lactose adsorption on the exposed crystal facets of metals: a comparative study of gold, silver and copper
N. Tarrat, J. C. Schön, and J. Cortés
Physical Chemistry Chemical Physics 26 (2024) 21134
DOI: https://doi.org/10.1039/D4CP01559B
IGLOO: an Iterative Global exploration and LOcal Optimization algorithm to find diverse low-energy conformations of flexible molecules
W. Margerit, A. Charpentier, C. Maugis-Rabusseau, J. C. Schön, N. Tarrat, and J. Cortés
Algorithms 16 (2023) 476
DOI: https://doi.org/10.3390/a16100476