Our research is multidisciplinary (physics, chemistry, biology) and concerns the elaboration and study of the structural, physical and chemical properties of i) surfaces and planar interfaces and grain boundaries, ii) nano-objects and iii) their interactions with atoms or molecules (adsorption, surface self-organization, ligand-directed growth and functionalization) (see diagram below). Our field of research on these objects, whether 2D (surfaces and planar interfaces and grain boundaries) or 3D (nano-objects), covers areas ranging from formation mechanisms (nucleation, epitaxy, wetting, elasticity, diffusion and alloying effects, kinetics, role of ligands, a gaseous environment or a matrix…) to physical properties (structural, mechanical, magnetic, electronic, optical…) or chemical properties (reactivity, bio-activity…). Our research is fundamental, however a large part of our objects of study are derived from functional devices or aiming to become so, such as lasers, devices for spintronics or nanoparticles for hyperthermia, antibiotherapy or catalysis. An originality of the SINanO group is to couple experimental and theoretical approaches on a large number of subjects.

Keywords: nanoparticles, single objects, crystalline surfaces and interfaces, hetero-epitaxy, atomic structure, surface self-organization, self-assembly, surface-ligand interaction, bioactivity, growth mechanisms, nucleation, out-of-equilibrium systems, metastability
Tools and methods: ultra-high vacuum processing (sputtering and cluster source); atomic scale analysis: structural and chemical (HRTEM, STEM-HAADF, EDX, …) and electronic (STM-BT); magnetic measurements; theory; classical, quasi-ab-initio and ab-initio modelling, molecular dynamics, machine learning
Group leader: Magali Benoit
Co-animation: Anne Ponchet & Roland Coratger
Selection of our most recent publications on this theme (from 2018) – Team members’ names are in blue.
_______________________________________________
Morphology and symmetry driven by lattice accommodation in polycrystalline bcc-fcc core-shell metallic nanoparticles
A. Ponchet, N. Tarrat, T. Hungria, M. Benoit, M.-J. Casanove, and P. Benzo
J. Appl. Phys. , 2023, 134, 205301
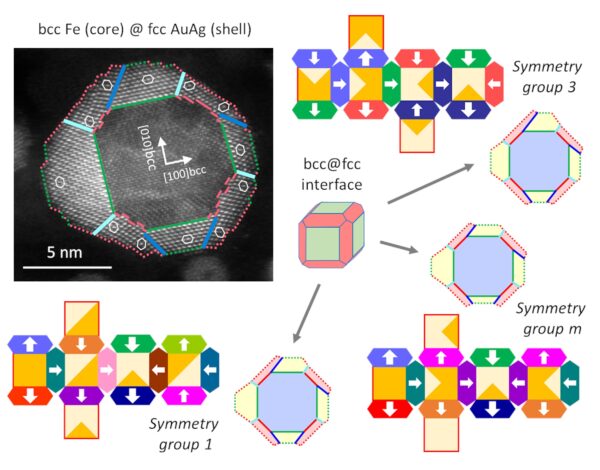
Body-centered-cubic–face-centered-cubic (bcc–fcc) multi-metallic nanoparticles (NPs) associating a single-crystal core (Fe, FeCo alloys, etc.) with a polycrystalline noble metal shell (Au, AuAg alloys, etc.) are perfectly symmetrical or more irregular, even dramatically dissymmetrical, yet presenting a good crystalline organization. Here, a combination of experimental analysis and theoretical symmetry analysis is proposed, in order to provide a unified description of the observed morphologies (Fe–Au and Fe–AuAg systems), whatever their symmetry, and predict some morphology variability in a population of NPs. First, the central role of the crystal lattice accommodation is comprehensively analyzed from the experimental Fe–AuAg system. The two possible bcc–fcc epitaxial relationships generate a core–shell interface in the shape of a truncated rhombic dodecahedron. This results in two different types of grains in the shell, which are elastically accommodated between them by an equal distribution of twins and low-angle grain boundaries, however, at the cost of internal stresses. At the same time, symmetry breaking results from two possible growth variants originating from the Nishiyama–Wasserman epitaxial relationships. The shell grains fit together in a nanopuzzle-like organization, resulting in a large number of possible arrangements distributed in 13 different point groups of symmetry, all of lower order than the core symmetry (highest order of cubic symmetry). If the variants are randomly distributed, the probability for the NP to be asymmetric (group 1) is 80%. The dissymmetrical development of the NPs is then discussed. Extending this approach to other core shapes succeeds in predicting dissymmetrical or dramatically off-centered morphologies experimentally observed in Fe–Au NPs.
_______________________________________________
Morphological sensitivity of silver nanoparticles to the environment
N. Tarrat and D. Loffreda
Environmental Science: Nano, 2023, 10, 1754

Silver nanoparticles have been modeled by density functional theory calculations and ab initio molecular dynamics simulations to explore their stability in air at room temperature. The role of contaminants is considered by adsorbing monoshells of methanethiol molecules on silver nanoparticles of various morphologies (icosahedral, strongly irregular and regular truncated octahedral structures) in the range of 147–201 atoms (∅ = 1.66–1.80 nm), under atmospheric nitrogen pressure. While ab initio molecular dynamics simulations suggest that the icosahedral and all the face-centered cubic FCC clusters are stable at 300 K under vacuum, the ino-decahedral geometry progressively transforms into a complex structure composed of an irregularly icosahedral outershell and a decahedral core. Nanoparticle surface energies, computed at 0 K and 300 K, show a preference for FCC clusters in vacuum, as previously reported experimentally. In the presence of air at 300 K, the icosahedral cluster presents the largest exothermicity in terms of adsorption surface energy of contaminant monoshells. This energetic gain is understood on the basis of a larger surface silver atomic density for the icosahedral structure, which better accommodates dense contaminant monoshells than FCC clusters. The methanethiol adlayers are composed of a complex phase of chemisorbed molecules bound to silver and in interaction with physisorbed contaminants through hydrogen bonds. This theoretical study agrees with measurements of silver nanoparticles exposed to air after synthesis in vacuum and also investigated in solution, and demonstrates that the air environment tunes the relative stability of morphologies in competition. This work paves the way for the understanding of nanoparticle ageing in environmental conditions at the atomic scale.
_______________________________________________
Influence of Air Exposure on Structural Isomers of Silver Nanoparticles
J. Vernieres, N. Tarrat, S. Lethbridge, E. Watchorn-Rokutan, T. Slater, D. Loffreda, and R. E. Palmer
Communications Chemistry, 2023, 6, 19

Up to date, the influence of ambient air exposure on the energetics and stability of silver clusters has rarely been investigated and compared to clusters in vacuum. Silver clusters up to 3000 atoms in size, on an amorphous carbon film, have been exposed to ambient air and investigated by atomic-resolution imaging in the aberration-corrected Scanning Transmission Electron Microscope. Ordered structures comprise more than half the population, the rest are amorphous. Here, we show that the most common ordered isomer structures is the icosahedron. These results contrast with the published behaviour of silver clusters protected from atmospheric exposure, where the predominant ordered isomer is face-centred cubic. We propose that the formation of surface oxide or sulphide species resulting from air exposure can account for this deviation in stable isomer. This interpretation is consistent with density functional theory calculations based on silver nanoclusters, in the size range 147-201 atoms, on which methanethiol molecules are adsorbed. An understanding of the effects of ambient exposure on the atomic structure and therefore functional properties of nanoparticles is highly relevant to their real-world performance and applications.
_______________________________________________
Silica-induced electron loss of silver nanoparticles
M.Benoit, J. Puibasset, C. Bonafos and N. Tarrat
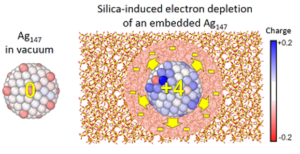
Despite the frequent use of silver nanoparticles (Ag NP) embedded in
materials for medical or optical applications, the effect of the matrix
on the nanoparticles properties remains largely unknown. This study aims
to shed light on the effect of an amorphous silica matrix on the
structure and charge distribution of 55- and 147-atoms silver
nanoparticles by means of dispersion-corrected DFT calculations.
Particular attention is paid on nanoparticle size and concentration
effects and on the impact of the presence of native defects in the
matrix. Covalent bonding between the silver nanoparticles and the matrix
are found to occur at the interface. Such interface reconstruction
involves the breaking of Si-O bonds, which systematically leads to the
formation of Ag-Si bonds, and in some cases, to the formation of Ag-O
ones. Interestingly, these interface reconstructions are accompanied by
an electron depletion of the nanoparticle, a substantial number of
electrons being transferred from the two outer shells of the Ag NP
towards the surrounding silica medium. The electrons lost by the
nanoparticle are captured by the Si atoms involved in the interface
bonds, but also, unexpectedly, by the undercoordinated silica defects
that act as electron pumps and by the atoms of the silica network inside
a spherical shell of a few angströms around the silver nanoparticle.
The number of interface bonds and of electrons transferred to the
surrounding silica shell appears to be proportional to the surface area
of the Ag NP. The electronic extension within the silica goes beyond
that attributable to the Ag NP spill-out. The presence of additional
electrons in the matrix, especially on defects, is consistent with the
experimental literature.
_______________________________________________
Periodic DFTB for supported clusters: Implementation and application to benzene dimers deposited on graphene
Mathias Rapacioli and Nathalie Tarrat
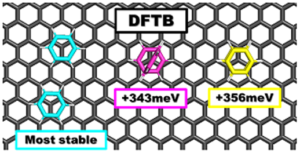
The interest for properties of clusters deposited on surfaces has grown in recent years. In this framework, the Density Functional based Tight Binding (DFTB) method appears as a promising tool due to its ability to treat extended systems at the quantum level with a low computational cost. We report the implementation of periodic boundary conditions for DFTB within the deMonNano code with k-points formalism and corrections for intermolecular interactions. The quality of DFTB results is evaluated by comparison with dispersion-corrected DFT calculations. Optimized lattice properties for a graphene sheet and graphite bulk are in agreement with reference data. The deposition of both benzene monomer and dimers on graphene are investigated and the observed trends are similar at the DFT and DFTB levels. Moreover, interaction energies are of similar orders of magnitude for these two levels of calculation. This study has evidenced the high stability of a structure made of two benzene molecules deposited close to each other on the graphene sheet. This work demonstrates the ability of the new implementation to investigate surface-deposited molecular clusters properties.
_______________________________________________
Measuring transferability issues in machine-learning force fields: The example of Gold-Iron interactions with linearized potentials
M.Benoit, J. Amodeo, S. Combettes, A. Roux, I. Khaled, J. Lam
Mach. Learn. : Sci. Technol. 2021, 2, 025003

De par leur capacité à faire le pont entre la précision des modèles basés sur la structure électronique et la rapidité des potentiels d’interactions classiques, les méthodes machine-learning ont été largement plébiscitées pour le développement de champs de forces entre les atomes. Néanmoins, un inconvénient de ces nouvelles méthodes demeure leur possible incapacité à extrapoler au-delà de la base de données utilisée pendant la phase d’apprentissage.
Dans cet article, nous avons commencé par présenter une nouvelle méthode machine-learning basée sur l’utilisation d’un algorithme de régression linéaire sous contrainte et nous avons montré qu’il était alors possible de construire des champs de forces pour des nanoparticules Fe/Au tout en contrôlant à la fois la complexité et la précision du potentiel. Ensuite, en testant les potentiels obtenus sur des structures non explorées dans la base de données d’apprentissage, nous avons observé un surprenant caractère non-monotone de la précision vis à vis de la complexité.
Ces résultats montrent de manière quantitative une limitation des méthodes machine-learning et tend ainsi vers une meilleure compréhension de leur utilisation.
_______________________________________________
Plasmon damping and charge transfer pathways in Au@MoSe2 Nanostructures
I.Abid, P. Benzo, B. Pecassou, S. Jia, J. Zhang, J. Yuan, J.B. Dory, O. Gauthier Lafaye, R. Pechou, A. Mlayah, J. Lou
Materials Today Nano 2021, 15, 100131

(A) Cross-section drawing of the sample, (B) optical microscopy image, (C) SEM, and (D) AFM topography images of an MoSe2 flake covered by Au NPs deposited with 4.8-nm Au equivalent thickness. (E) SEM image of an MoSe2 flake covered by Au NPs deposited with 0.8-nm Au equivalent thickness.
Hybridization of plasmonic and excitonic elementary excitations provides an efficient mean of enhancing the optical absorption and emission properties of metal/semiconductor nanostructures and is a key concept for the design of novel efficient optoelectronic devices. Here we investigate the optical properties of two-dimensional MoSe2 quantum well flakes covered with Au nanoparticles supporting plasmonic resonances. Using spatially resolved confocal spectroscopy, we report the observation of a quenching phenomenon of the Raman scattering and photoluminescence emission of both the MoSe2 layer and the Au nanoparticles. We found that the quenching of the photoluminescence emission from the Au nanoparticles is partial and measurable unlike the one observed for the Au-covered MoSe2 layers, which is total. Its dependence on the thickness of the MoSe2 layer is determined experimentally. Based on electrodynamics calculations and on the electronic band alignment at the Au/MoSe2 interface, the results are interpreted in terms of (1) damping of the plasmonic resonance of the Au nanoparticles due to the optical absorption by the MoSe2 layer and (2) a two-pathways charge transfer scheme where the photoexcited electrons leak from the MoSe2 layer to the Au NPs, whereas the photoexcited holes flow in the opposite direction, that is, from the Au NPs to the MoSe2 layer. The two combined mechanisms account well for the experimental observations and complements the interpretations proposed in the literature for similar metal nanoparticles/transition metal dichalcogenide systems.
_______________________________________________
Exploring energy landscapes at the DFTB quantum level using the threshold algorithm: the case of the anionic metal cluster Au20–
M. Rapacioli, J. C. Schoen and N. Tarrat
Theor. Chem. Acc. 2021, 140, 85
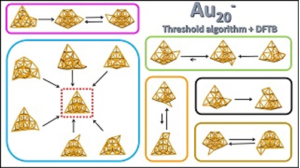
We report the combination of the threshold algorithm with the Density Functional-based Tight Binding method allowing for the exploration of complex potential energy surfaces and the evaluation of probability flows between their regions, at the quantum level. This original scheme is used to explore the energy landscape of an anionic 20-atom gold cluster, Au−20. On the basis of the relevant structures, 19 structural groups are highlighted, all of them being variations about the pyramidal shape: (1) distorted pyramids, (2) pyramids in which the atom of one of the facets has been removed, leaving a hole, and placed at different positions on the cluster and (3) pyramids on which an atom located at a vertex has been removed and placed on an edge or on a facet. Upper limits of the energies required to connect the basins of the 19 groups on the potential energy surface are evaluated. Moreover, the attractive basins are identified on the basis of the analysis of the probability flows on the landscape. The comparison of the disconnectivity tree with the results of the flux analysis provides a consistent representation of the Au basins’ proximity. Finally, we show how the new scheme allowed for the identification of counter-intuitive transition pathways.
_______________________________________________
Importance of Defective and Nonsymmetric Structures in Silver Nanoparticles
David Loffreda, Dawn M. Foster, Richard E. Palmer, and Nathalie Tarrat
Phys. Chem. Lett. 2021, 12, 3705
 Scanning transmission electron microscopy experiments indicate that face-centered cubic (FCC) is the predominant ordered structure for Ag309 ± 7 nanoclusters, synthesized in vacuum. Historically, experiments do not present a consensus on the morphology at these sizes, whereas theoretical studies find the icosahedral symmetry for Ag309 and the decahedral shape for nearby sizes. We employ density functional theory calculations to rationalize these observations, considering both regular and defective Ag nanoparticles (281−321 atoms). The change of stability induced by the presence of defects, symmetry loss, and change of number of atoms is evaluated by the nanoparticle surface energy, which was measured previously. FCC and decahedral symmetries are found to be more favorable than icosahedral, consistent with our measurements of clusters protected from extended atmospheric exposure. In addition, an energy-free descriptor, surface atomic density, is proposed and qualitatively reproduces the surface energy data. Nonsymmetric and defective structures may be preferred over perfectly regular ones within a given size range.
Scanning transmission electron microscopy experiments indicate that face-centered cubic (FCC) is the predominant ordered structure for Ag309 ± 7 nanoclusters, synthesized in vacuum. Historically, experiments do not present a consensus on the morphology at these sizes, whereas theoretical studies find the icosahedral symmetry for Ag309 and the decahedral shape for nearby sizes. We employ density functional theory calculations to rationalize these observations, considering both regular and defective Ag nanoparticles (281−321 atoms). The change of stability induced by the presence of defects, symmetry loss, and change of number of atoms is evaluated by the nanoparticle surface energy, which was measured previously. FCC and decahedral symmetries are found to be more favorable than icosahedral, consistent with our measurements of clusters protected from extended atmospheric exposure. In addition, an energy-free descriptor, surface atomic density, is proposed and qualitatively reproduces the surface energy data. Nonsymmetric and defective structures may be preferred over perfectly regular ones within a given size range.
_______________________________________________
How interface properties control the equilibrium shape of core–shell Fe–Au and Fe–Ag nanoparticles
Ségolène Combettes, Julien Lam, Patrizio Benzo, Anne Ponchet, Marie-José Casanove, Florent Calvo and Magali Benoit
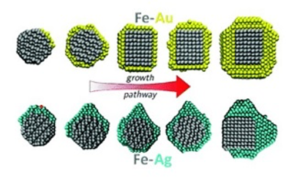
Growth simulations of Fe-Au and Fe-Ag nanoparticles
While combining two metals in the same nanoparticle can lead to remarkable novel applications, the resulting structure in terms of crystallinity and shape remains difficult to predict. It is thus essential to provide a detailed atomistic picture of the underlying growth processes. In the present work we address the case of core–shell Fe–Au and Fe–Ag nanoparticles. Interface properties between Fe and the noble metals Au and Ag, computed using DFT, were used to parameterize Fe–Au and Fe–Ag pairwise interactions in combination with available many-body potentials for the pure elements. The growth of Au or Ag shells on nanometric Fe cores with prescribed shapes was then modelled by means of Monte Carlo simulations. The shape of the obtained Fe–Au nanoparticles is found to strongly evolve with the amount of metal deposited on the Fe core, a transition from the polyhedral Wulff shape of bare iron to a cubic shape taking place as the amount of deposited gold exceeds two monolayers. In striking contrast, the growth of silver proceeds in a much more anisotropic, Janus-like way and with a lesser dependence on the iron core shape. In both cases, the predicted morphologies are found to be in good agreement with experimental observations in which the nanoparticles are grown by physical deposition methods. Understanding the origin of these differences, which can be traced back to subtle variations in the electronic structure of the Au/Fe and Ag/Fe interfaces, should further contribute to the better design of core–shell bimetallic nanoparticles.
_______________________________________________
Equilibrium shape of core(Fe)–shell(Au) nanoparticles as a function of the metals volume ratio
A.Ponchet, S. Combettes, P. Benzo, N. Tarrat, M. J. Casanove, and M. Benoit
Journal of Applied Physics 2020, 128, 055307
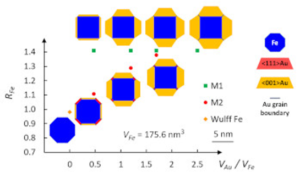
The equilibrium shape of nanoparticles is investigated to elucidate the various core–shell morphologies observed in a bimetallic system associating two immiscible metals, iron and gold, that crystallize in the bcc and fcc lattices, respectively. Fe–Au core–shell nanoparticles present a crystalline Fe core embedded in a polycrystalline Au shell, with core and shell morphologies both depending on the Au/Fe volume ratio. A model is proposed to calculate the energy of these nanoparticles as a function of the Fe volume, Au/Fe volume ratio, and the core and shell shape, using the density functional theory-computed energy densities of the metal surfaces and of the two possible Au/Fe interfaces. Three driving forces leading to equilibrium shapes were identified: the strong adhesion of Au on Fe, the minimization of the Au/Fe interface energy that promotes one of the two possible interface types, and the Au surface energy minimization that promotes a 2D–3D Stranski–Krastanov-like transition of the shell. For a low Au/Fe volume ratio, the wetting is the dominant driving force and leads to the same polyhedral shape for the core and the shell, with an octagonal section. For a large Au/Fe ratio, the surface and interface energy minimizations can act independently to form an almost cube-shaped Fe core surrounded by six Au pyramids. The experimental nanoparticle shapes are well reproduced by the model, for both low and large Au/Fe volume ratios. J. Appl. Phys. 128, 055307 (2020)
_______________________________________________
Epitaxial growth of a gold shell on intermetallic FeRh nanocrystals
P.Benzo, S. Combettes, C. Garcia, T. Hungria, B. Pecassou, and M. J. Casanove
Cryst. Growth Des. 2020, 20, 6, 4144

In contrast with bimetallics, multimetallic nanoparticles (NPs) can combine different chemical orders in a same NP, which favors an enhanced tuning of their properties. In this work, trimetallic (Fe,Rh,Au) nanocrystals with controlled composition and chemical distribution were grown through a physical vapor deposition route using a two-step process. First FeRh nanocrystals, 8.5 nm of mean diameter, were formed according to a Volmer-Weber growth mode. The growth conditions were tuned so as to achieve the atomic scale chemical order displayed by the intermetallic B2-FeRh phase. Then the gold layer was deposited at a lower temperature. Evidence is given for the complete coverage of the gold shell, which grows epitaxially over the B2-FeRh core exposed facets. These different features are particularly promising for further applications.
_______________________________________________
Role of the shell thickness in the core transformation of magnetic core(Fe)-shell(Au) nanoparticles
P. Benzo, S. Combettes, B. Pecassou, N. Combe, M. Benoit, M. Respaud, M.J. Casanove
Phys. Rev. Materials 2019, 3, 096001
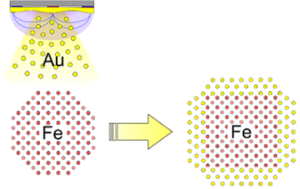
Fe-Au core-shell nanoparticles embedded in an amorphous alumina matrix are synthesized at high temperature using magnetron sputtering. The nanoparticles display a single-crystal Fe core covered by a crystalline Au shell epitaxially grown on the different core facets. The morphology of the grown nanoparticles is analyzed by transmission electron microscopy, while their structural details are resolved at the atomic scale using probe-corrected high-angle annular dark-field scanning transmission microscopy. Two different epitaxial relationships at the Au/Fe interface are observed at low Au coverage, whereas only one type of interface orientation remains when the shell thickness exceeds 3 monolayers (MLs). This leads to a drastic Fe core transformation from a Wulff shaped crystal towards a nanocube. This core transformation drives a surface reconstruction resulting in a combination of open and close-packed strain free facets, offering different opportunities for molecule binding. In addition, our experiments show that the magnetic properties of the Fe core are preserved by the Au shell and that the 10 nm size of the grown nanoparticles favors a superparamagnetic behavior, suitable for biomedical applications.
_______________________________________________
Au147 nanoparticles : Ordered or amorphous ?
Nathalie Tarrat, Mathias Rapacioli, Fernand Spiegelman
J. Chem. Phys. 2018, 148, 204308
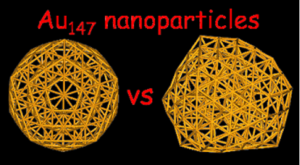
Structural aspects of the Au147 nanoparticles have been investigated through a density functional based tight binding global optimization involving a parallel tempering molecular dynamics scheme with quenching followed by geometries relaxation at the Density Functional Theory (DFT) level. The focus is put on the competition between relaxed ordered regular geometries and disordered (or amorphous) structures. The present work shows that Au147 amorphous geometries are relevant low energy candidates and are likely to contribute in finite temperature dynamics and thermodynamics. The structure of the amorphous-like isomers is discussed from the anisotropy parameters, the atomic coordinations, the radial and pair distribution functions, the IR spectra, and the vibrational DOS. With respect to the regular structures, the amorphous geometries are shown to be characterized by a larger number of surface atoms, a less dense volume with reduced coordination number per atom, a propensity to increase the dimension of flat facets at the surface, and a stronger anisotropy. Moreover, all amorphous clusters have similar IR spectra, almost continuous with active frequencies over the whole spectral range, while symmetric clusters are characterized by a few lines with large intensities.
Selection of our most recent publications on this theme (from 2018) – Team members’ names are in blue.
_______________________________________________
Evidencing the relationship between isomer spectra and melting: the 20- and 55-atom silver and gold cluster cases
Mathias Rapacioli, Fernand Spiegelman and Nathalie Tarrat
Phys. Chem. Chem. Phys. 2019, 21, 24857
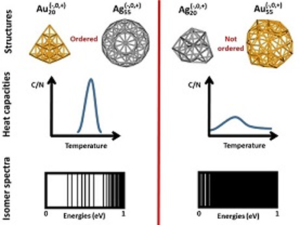 The present work highlights the links between melting properties and structural excitation spectra of small gold and silver clusters. The heat capacity curves are computed for Ag20, Au20, Ag55, Au55 and their ions, using a parallel-tempering molecular dynamics scheme to explore the density functional based tight binding (DFTB) potential energy surfaces and the multiple histogram method. It is found that clusters having very symmetric lowest energy structures (Au20, Ag55 and their ions) present sharp or relatively sharp solid-to-liquid transitions and large melting temperatures, important structural excitation energies and a discrete isomer spectrum. Opposite trends are observed for less ordered clusters (Ag20, Au55 and their ions). Regarding the structural evolution with temperature, very symmetric clusters exhibit minor evolution up to the starting melting temperature. The present study also highlights that, in contrast with the case of Au20, a single electron excess or deficiency is not determinant regarding the melting characteristics, even quantitatively, for clusters containing 55 atoms, for gold as for silver.
The present work highlights the links between melting properties and structural excitation spectra of small gold and silver clusters. The heat capacity curves are computed for Ag20, Au20, Ag55, Au55 and their ions, using a parallel-tempering molecular dynamics scheme to explore the density functional based tight binding (DFTB) potential energy surfaces and the multiple histogram method. It is found that clusters having very symmetric lowest energy structures (Au20, Ag55 and their ions) present sharp or relatively sharp solid-to-liquid transitions and large melting temperatures, important structural excitation energies and a discrete isomer spectrum. Opposite trends are observed for less ordered clusters (Ag20, Au55 and their ions). Regarding the structural evolution with temperature, very symmetric clusters exhibit minor evolution up to the starting melting temperature. The present study also highlights that, in contrast with the case of Au20, a single electron excess or deficiency is not determinant regarding the melting characteristics, even quantitatively, for clusters containing 55 atoms, for gold as for silver.
_______________________________________________
Melting of the Au20 Gold Cluster: Does Charge Matter?
Mathias Rapacioli, Nathalie Tarrat, Fernand Spiegelman
J. Phys. Chem. A 2018, 122, 16, 4092

We investigate the dependence upon the charge of the heat capacities of magic gold cluster Au20 obtained from density functional-based tight binding theory within parallel tempering molecular dynamics and the multiple histogram method. The melting temperatures, determined from the heat capacity curves, are found to be 1102 K for neutral Au20 and only 866 and 826 K for Au20+ and Au20–. Both the canonical and the microcanonical aspects of the transition are discussed. A convex intruder, associated with a negative heat capacity, is evidenced in the microcanonical entropy in the case of the neutral cluster but is absent in the melting processes of the ions. The present work shows that a single charge quantitatively affects the thermal properties of the gold 20mer.
Selection of our most recent publications on this theme (from 2018) – Team members’ names are in blue.
_______________________________________________
Multiple coupling modes to relax shear strain during grain boundary migration
Combe, N.; Mompiou, F. & Legros, M.
Acta Materialia 2021, 218, 117222

Shear-coupled grain boundary (GB) migration is an effective plastic mechanism in absence of dislocation activity, ie. more favorably in nanocrystalline metals. For a given GB, several stress induced migration mechanisms, referred as coupling modes participate to the decrease of the elastic energy produced by the shear. They operate through the nucleation and motion of interfacial defects known as disconnections, carrying elementary shear strain characterized by their Burgers vector. However, so far, the coupling modes have been studied only under a simple shear, a situation much less complex than expected in a strained polycrystal, where multiple components of the stress tensor are present. Here we propose a more systematic investigation of the coupling modes when a composite shear is applied. This promotes the activation of new coupling modes. Using Molecular Dynamics simulations, we evidence these multiple coupling modes and the operation of their associate disconnections. Moreover, we also show that, even at low temperature, GB migration may occur by the successive occurrence of two modes : the relaxed shear appears then as an effective parameter, resulting from the combination of two operating elementary mechanisms.
_______________________________________________
Migration coupled to grain boundary shear: the missing link in the mechanical behavior of small-grained metals?
Gautier, R.; Rajabzadeh, A.; Larranaga, M.; Combe, N.; Mompiou, F. & Legros, M.
Comptes Rendus. Physique, Académie des sciences, Paris, 2021, 22, 19-34
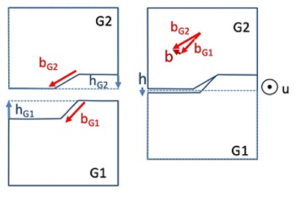
Grain size reduction is a very effective way to block dislocation movements and thus increase the mechanical strength of metals and alloys. Not only are grain boundaries known obstacles for dislocations, but when they reach nanometer dimensions, crystallites typically become empty of dislocations, imposing additional stress to develop plasticity. Understanding the deformation mechanisms based on grain boundaries has become a major issue in physical metallurgy. These mechanisms can be multiple, involving conservative and diffusive processes that are poorly understood. A first approach, which consists in transposing to small dimensions mechanisms documented on a large scale such as Coble creep, has proven to be very limited. Instead, stress-assisted grain growth or shear-coupled grain boundary migration, recently observed in small-grained materials at room temperature, may provide a key to fully understand “dislocation-free plasticity” in nanocrystals. As this is a relatively new field with many more degrees of freedom, a continued research effort must be conducted to link the mechanical properties of nanocrystals to these grain boundary-based plasticity processes.
_______________________________________________
Zinc-blende group III-V/group IV epitaxy: Importance of the miscut
Cornet, S. Charbonnier, I. Lucci, L. Chen, A. Létoublon, A. Alvarez, K. Tavernier, T. Rohel, R. Bernard, J.-B. Rodriguez, L. Cerutti, E. Tournié, Y. Léger, M. Bahri, G. Patriarche, L. Largeau, A. Ponchet, P. Turban, and N. Bertru
Phys. Rev. Materials 2020, 4, 053401
We clarify the central role of the miscut during group III-V/group IV crystal growth. We show that the miscut impacts the initial antiphase domain distribution, with two distinct nucleation-driven (miscut typically >1°) and terraces-driven (miscut typically <0.1°) regimes. It is then inferred how the antiphase domain distribution mean phase and mean lateral length are affected by the miscut. An experimental confirmation is given through the comparison of antiphase domain distributions in GaP and GaSb/AlSb samples grown on nominal and vicinal Si substrates. The antiphase domain burying step of GaP/Si samples is then observed at the atomic scale by scanning tunneling microscopy. The steps arising from the miscut allow growth rate imbalance between the two phases of the crystal and the growth conditions can deeply modify the imbalance coefficient, as illustrated with GaAs/Si. We finally explain how a monodomain III-V semiconductor configuration can be achieved even on low miscut substrates. Phys. Rev. Materials 4, 053401 (2020).
_______________________________________________
Density Functional Theory Study of the Spontaneous Formation of Covalent Bonds at the Silver/Silica Interface in Silver Nanoparticles Embedded in SiO2: Implications for Ag+ Release
Hilal Balout, Nathalie Tarrat, Joël Puibasset, Simona Ispas, Caroline Bonafos, Magali Benoit
ACS Appl. Nano Mater. 2019, 2, 8, 5179–5189

Silver nanoparticles (AgNPs) are widely used in the health-care sector and industrial applications because of their outstanding antibacterial activity. This bactericidal effect is mainly attributed to the release of Ag+ ions in an aqueous medium, the first step of which is the dissolution of the AgNP via the oxidation of its surface by O2. With the aim of designing more durable and less toxic antibacterial devices, it is desirable to fine-tune the rate of Ag+ release into the surrounding environment. This can be achieved by choosing an adequate coating of the AgNPs, e.g., by embedding the nanoparticles in a silica matrix. In a previous work (Pugliara, A. ; et al. Sci. Total Environ.2016, 565, 863), we have shown that the toxic effect on algae photosynthesis of small AgNPs (size <20 nm) embedded in silica layers is preserved, provided that the distance between the AgNPs and the silica free surface is below ≈6–7 nm. Better control of the Ag+ release rate in these systems requires a better understanding of the elementary mechanisms at play concerning both the detachment of the Ag ions from the AgNPs and their diffusion through SiO2. A first step in this direction consists in characterizing the interface between the AgNPs surface and the silica matrix. In this context, periodic density functional theory calculations have been performed on model systems representing the interfaces between amorphous silica and the three crystalline facets of AgNPs, i.e., Ag(111), Ag(110), and Ag(100). Spontaneous breaking of the Si–O bonds and the formation of two O–Ag and one Si–Ag bonds are observed in 50% of the investigated interfaces, corresponding to 1.8 bonds/nm2 on average. The covalent nature of the bonds between Ag and O and between Ag and Si is highlighted by analysis of the electronic structure of the interfaces.
_______________________________________________
Heterogeneous disconnection nucleation mechanisms during grain boundary migration
N.Combe, F. Mompiou, and M. Legros
Phys. Rev. Materials 2019 3, 060601(R)
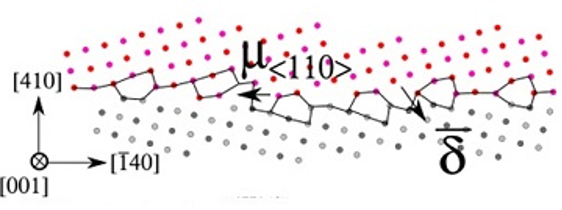
Heterogeneous nucleation of a mobile μ⟨110⟩ disconnection from a sessile disconnection
Shear-coupled grain boundary (GB) migration has been evidenced as an efficient mechanism of plasticity in the absence of dislocation activity. The GB migration occurs through the nucleation and motion of disconnections. Using molecular simulations, we report a detailed study of the elementary mechanisms occurring during heterogeneous disconnection nucleation. We study the effect of a pre-existing sessile disconnection in asymmetric S17(410) [001] tilt GB on the GB migration mechanism. Shearing this imperfect GB induces its migration and reveals a new GB migration mechanism through the nucleation of a mobile disconnection from the sessile one. Energy barriers and yield stress for the GB migrations are evaluated and compared to the migration of a perfect GB. We show that the migration of the imperfect GB is easier than the perfect one and that a sessile disconnection can operate as a source of disconnection driving the GB migration. This GB migration mechanism has been observed on two other high-angle GBs.
_______________________________________________
Shape transition in InAs nanostructures formed by Stranski-Krastanow growth mode on InP (001) substrate
A. Ponchet, L. Pedesseau, A. Le Corre, C. Cornet, and N. Bertru
Appl. Phys. Lett. 2019, 114, 173102
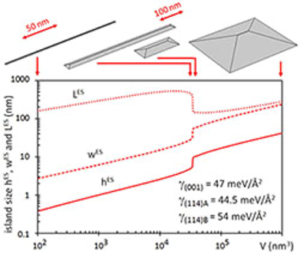
Equilibrium shape (length, width and height) as a function of the volume of InAs nanostructures grown on InP(001) substrates
The shape of InAs nanostructures formed by molecular beam epitaxy on a (001) InP substrate in the Stranski-Krastanow growth mode is studied. A transition from wires to round-shaped islands is observed as a function of the amount of InAs deposited. It is attributed to the non-equivalent energies of the A and B facets existing in zinc blende materials (facets along [1-10] and [110], respectively). This surface energy anisotropy is considered to determine the nanostructure equilibrium shape from the balance between the elastic energy and the surface energy. At low volumes, the most energetically favorable shape is the wire-like shape, while at high volumes, the equilibrium shape is the island-like shape. The calculated sizes for which the shape changes are in good agreement with experimental sizes. The low lattice mismatch and the low surface energy of (114)A InAs facets around 41 meV/A2, as obtained from density functional theory calculations, enhance this effect in the InAs/InP system. Published in Appl. Phys. Lett. 114, 173102 (2019).
_______________________________________________
Interface energy analysis of III–V islands on Si (001) in the Volmer-Weber growth mode
A. Ponchet, G. Patriarche, J. B. Rodriguez, L. Cerutti and E. Tournié
Appl. Phys. Lett. 2018, 113, 191601

The in-plane shape of III-V on Si islands varies from quasi rounded to elongate depending on whether the adhesion is low or high.
The experimental island shapes of III–V islands grown on silicon (001) in the Volmer-Weber growth mode are analyzed in the frame of the theory of wetting in crystals. A reverse Wulff-Kaishew (or Winterbottom) construction is used in order to access interfacial energy. We apply this approach to AlSb and GaSb islands on (001) Si grown by molecular beam epitaxy and observed by scanning transmission electron microscopy. Experimental ratios between energies of (001), (110), (111)A, and (111)B surfaces are established. Interface energies are then quantitatively estimated for GaSb/Si and AlSb/Si interfaces. The differences in the shape of GaSb and AlSb islands, which are consistently reported in the literature, can be clearly attributed to a higher energy for the GaSb/Si interface compared to the ASb/Si one and not to different adatom diffusion lengths. The difference in interface energies is quantified, and its origin at the microscopic level is discussed. Published in Appl. Phys. Lett. 113, 191601 (2018).
_______________________________________________
A Stress-Free and Textured GaP Template on Silicon for Solar Water Splitting
Ida Lucci, Simon Charbonnier, Maxime Vallet, Pascal Turban, Yoan Léger, Tony Rohel, Nicolas Bertru, Antoine Létoublon, Jean-Baptiste Rodriguez, Laurent Cerutti, Eric Tournié, Anne Ponchet, Gilles Patriarche, Laurent Pedesseau and Charles Cornet
Adv. Funct. Mater. 2018, 28, 1801585
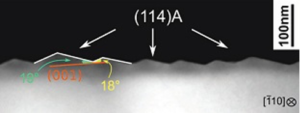 This work shows that a large-scale textured GaP template monolithically integrated on Si can be developed by using surface energy engineering, for watersplitting applications. The stability of (114)A facets is first shown, based on scanning tunneling microscopy images, transmission electron microscopy, and atomic force microscopy. These observations are then discussed in terms of thermodynamics through density functional theory calculations. A stress free nanopatterned surface is obtained by molecular beam epitaxy, composed of a regular array of GaP (114)A facets over a 2 in. vicinal Si substrate. The advantages of such textured (114)A GaP/Si template in terms of surface gain, band lineups, and ohmic contacts for water-splitting applications are finally discussed. Published in Adv. Funct. Mater. 2018, 1801585.
This work shows that a large-scale textured GaP template monolithically integrated on Si can be developed by using surface energy engineering, for watersplitting applications. The stability of (114)A facets is first shown, based on scanning tunneling microscopy images, transmission electron microscopy, and atomic force microscopy. These observations are then discussed in terms of thermodynamics through density functional theory calculations. A stress free nanopatterned surface is obtained by molecular beam epitaxy, composed of a regular array of GaP (114)A facets over a 2 in. vicinal Si substrate. The advantages of such textured (114)A GaP/Si template in terms of surface gain, band lineups, and ohmic contacts for water-splitting applications are finally discussed. Published in Adv. Funct. Mater. 2018, 1801585.
_______________________________________________
Universal description of III-V/Si epitaxial growth processes
Lucci, S. Charbonnier, L. Pedesseau, M. Vallet, L. Cerutti, J.-B. Rodriguez, E. Tournié, R. Bernard, A. Létoublon, N. Bertru, A. Le Corre, S. Rennesson, F. Semond, G. Patriarche, L. Largeau, P. Turban, A. Ponchet, and C. Cornet
Phys. Rev. Materials 2018, 2, 060401(R)

Prediction from surface and interface energies of the 3D growth mode of GaP on Si
Here, we experimentally and theoretically clarify III-V/Si crystal growth processes. Atomically resolved microscopy shows that monodomain three-dimensional islands are observed at the early stages of AlSb, AlN, and GaP epitaxy on Si, independently of misfit. It is also shown that complete III-V/Si wetting cannot be achieved in most III-V/Si systems. Surface/interface contributions to the free-energy variations are found to be prominent over strain relief processes. We finally propose a general and unified description of III-V/Si growth processes, including a description of the formation of antiphase boundaries. Published in PHYSICAL REVIEW MATERIALS 2, 060401(R) (2018)
_______________________________________________
Noble Metal Nanocluster Formation in Epitaxial Perovskite Thin Films
M. Lee, R. Arras, R. Takahashi, B. Warot-Fonrose, H. Daimon, M.J. Casanove, M. Lippmaa

We studied the synthesis of nanocomposite materials consisting of noble metal clusters embedded in an oxide semiconductor matrix. The embedded nanostructures form in a simple self-organized single-step growth process. The primary interest is in developing materials for photo-electrochemical energy conversion where spatially inhomogeneous band structures can enhance photogenerated charge separation and carrier extraction from a semiconductor. We show that spontaneous segregation of metallic Ir occurs during the initial growth of an Ir:SrTiO3 thin film. Cross-sectional transmission electron microscopy suggests that the nanoscale Ir clusters are epitaxial with the host lattice, and their presence is not detectable by surface morphology measurements. Published in ACS Omega 3 ,2169-2173 (2018).
Selection of our most recent publications on this theme (from 2018) – Team members’ names are in blue.
_______________________________________________
IGLOO: an Iterative Global exploration and LOcal Optimization algorithm to find diverse low-energy conformations of flexible molecules
W. Margerit, A. Charpentier, C. Maugis-Rabusseau, C. J. Schön, N. Tarrat and J. Cortés
The exploration of the energy landscape of a chemical system is essential for understanding and predicting its observable properties. In most cases, this is a challenging task due to the high complexity of such landscapes, which often consist of multiple, possibly hierarchical basins that are difficult to locate and thoroughly explore. In this study, we introduce a novel method, called IGLOO (Iterative Global Exploration and Local Optimization), which aims to achieve a more efficient global exploration of the conformational space compared to existing techniques. The method utilizes a tree-based exploration inspired by the Rapidly exploring Random Tree (RRT) algorithm originating from robotics. IGLOO dynamically adjusts its exploration strategy to both homogeneously scan the landscape and focus on promising regions, avoiding redundant exploration. We evaluated IGLOO using models of two polypeptides and compared its performance to the traditional basin-hopping method and a hybrid method that also incorporates the RRT algorithm. We find that IGLOO outperforms both alternative methods in terms of efficiently and comprehensively exploring the molecular conformational space. This approach can be easily generalized to other chemical systems such as molecules on surfaces or crystalline systems.
_______________________________________________
Light assisted synthesis of poly-para-phenylene on Ag(001)
V. Langlais, K. Schneider and H. Tang
Journal of Physics: Condensed Matter 2022, 34, 055001
A detailed study of poly-para-phenylene (PPP) obtained by light-assisted on-surface-synthesis (OSS) on Ag(100) was carried out by scanning tunneling microscopy and spectroscopy together with density functional theory calculations. The use of light in combination with heat allows to lower by 50 K annealing temperature the each stage of the Ullmann coupling. Debromination of the 4,4’’ dibromo-p-terphenyl precursors was thus realized at 300 K, the formation of the first oligomers from the organometallic intermediate by silver bridging atom release at 423 K and PPP by complete elimination of the silver at 473 K. This approach to lower the reaction temperature permits to enhance the Ag(100) surface reactivity to become comparable to that of Cu(111). The underlying mechanism of light effect was proposed to occur via surface mediated excitation, with the creation of photoexcited electrons known as hot electrons correlated with surface plasmon excitation. This original pathway combining both light and heat provides an additional parameter to control OSS by separating the precursor activation stage from the diffusion.
_______________________________________________
Coordination of Ethylamine on Small Silver Clusters: Structural and Topological (ELF, QTAIM) Analyses
Corinne Lacaze-Dufaure, Yann Bulteau, Nathalie Tarrat, David Loffreda, Pierre Fau, Katia Fajerwerg, Myrtil L. Kahn, Franck Rabilloud, Christine Lepetit
Inorganic Chemistry 2022, 61, 19, 7274
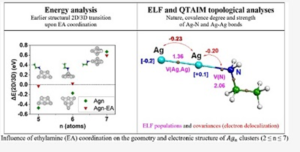 Amine ligands are expected to drive the organization of metallic centers as well as the chemical reactivity of silver clusters early growing during the very first steps of the synthesis of silver nanoparticles via an organometallic route. Density functional theory (DFT) computational studies have been performed to characterize the structure, the atomic charge distribution, and the planar two-dimensional (2D)/three-dimensional (3D) relative stability of small-size silver clusters (Agn, 2 ≤ n ≤ 7), with or without an ethylamine (EA) ligand coordinated to the Ag clusters. The transition from 2D to 3D structures is shifted from n = 7 to 6 in the presence of one EA coordinating ligand, and it is explained from the analysis of the Ag–N and Ag–Ag bond energies. For fully EA saturated silver clusters (Agn–EAn), the effect on the 2D/3D transition is even more pronounced with a shift between n = 4 and 5. Subsequent electron localization function (ELF) and quantum theory of atoms in molecules (QTAIM) topological analyses allow for the fine characterization of the dative Ag–N and metallic Ag–Ag bonds, both in nature and in strength. Electron transfer from ethylamine to the coordinated silver atoms induces an increase of the polarization of the metallic core.
Amine ligands are expected to drive the organization of metallic centers as well as the chemical reactivity of silver clusters early growing during the very first steps of the synthesis of silver nanoparticles via an organometallic route. Density functional theory (DFT) computational studies have been performed to characterize the structure, the atomic charge distribution, and the planar two-dimensional (2D)/three-dimensional (3D) relative stability of small-size silver clusters (Agn, 2 ≤ n ≤ 7), with or without an ethylamine (EA) ligand coordinated to the Ag clusters. The transition from 2D to 3D structures is shifted from n = 7 to 6 in the presence of one EA coordinating ligand, and it is explained from the analysis of the Ag–N and Ag–Ag bond energies. For fully EA saturated silver clusters (Agn–EAn), the effect on the 2D/3D transition is even more pronounced with a shift between n = 4 and 5. Subsequent electron localization function (ELF) and quantum theory of atoms in molecules (QTAIM) topological analyses allow for the fine characterization of the dative Ag–N and metallic Ag–Ag bonds, both in nature and in strength. Electron transfer from ethylamine to the coordinated silver atoms induces an increase of the polarization of the metallic core.
_______________________________________________
Adsorption of single metallic atoms on self-assembled molecular domain of terephthalic acid
H. Tang, C. Durand and R. Coratger
Surfaces and Interfaces 2021, 25, 101170
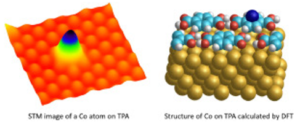 Cobalt and gold atoms have been deposited on self-assembled molecular domains of terephthalic acid and are studied using low temperature scanning tunneling microscopy and DFT calculations. These two types of atoms present different adsorption properties as well as peculiar electronic structures. Co atoms are firmly adsorbed on the molecules while Au is highly dependent of the presence of diffusing hydrogen atoms. These latter can also favour the possible desorption of the adatoms or change their adsorption site depending on the presence of defective molecules. The results open the way to new investigations concerning the use of single atoms or molecules for chemistry at the subnanometer scale.
Cobalt and gold atoms have been deposited on self-assembled molecular domains of terephthalic acid and are studied using low temperature scanning tunneling microscopy and DFT calculations. These two types of atoms present different adsorption properties as well as peculiar electronic structures. Co atoms are firmly adsorbed on the molecules while Au is highly dependent of the presence of diffusing hydrogen atoms. These latter can also favour the possible desorption of the adatoms or change their adsorption site depending on the presence of defective molecules. The results open the way to new investigations concerning the use of single atoms or molecules for chemistry at the subnanometer scale.
_______________________________________________
Integration of the Rhombohedral BiSb(0001) Topological Insulator on a Cubic GaAs(001) Substrate
D. SADEK, D.S. DHUNGANA, R. CORATGER, C. DURAND, A. PROIETTI, Q. GRAVELIER, B. REIG, E. DARAN, P.F. FAZZINI, F. CRISTIANO, A. ARNOULT, S.R. PLISSARD
ACS Applied Materials and Interfaces 2021, 13, 30, 36492
 Bismuth–antimony alloy (Bi1 – xSbx) is the first reported 3D topological insulator (TI). Among many TIs reported to date, it remains the most promising for spintronic applications thanks to its large conductivity, its colossal spin Hall angle, and the possibility to build low-current spin-orbit-torque magnetoresistive random access memories. Nevertheless, the 2D integration of TIs on industrial standards is lacking. In this work, we report the integration of high-quality rhombohedral BiSb(0001) topological insulators on a cubic GaAs(001) substrate. We demonstrate a clear epitaxial relationship at the interface, a fully relaxed TI layer, and the growth of a rhombohedral matrix on top of the cubic substrate. The antimony composition of the Bi1 – xSbx layer is perfectly controlled and covers almost the whole TI window. For optimized growth conditions, the sample generates a semiconductor band structure at room temperature in the bulk and exhibits metallic surface states at 77 K.
Bismuth–antimony alloy (Bi1 – xSbx) is the first reported 3D topological insulator (TI). Among many TIs reported to date, it remains the most promising for spintronic applications thanks to its large conductivity, its colossal spin Hall angle, and the possibility to build low-current spin-orbit-torque magnetoresistive random access memories. Nevertheless, the 2D integration of TIs on industrial standards is lacking. In this work, we report the integration of high-quality rhombohedral BiSb(0001) topological insulators on a cubic GaAs(001) substrate. We demonstrate a clear epitaxial relationship at the interface, a fully relaxed TI layer, and the growth of a rhombohedral matrix on top of the cubic substrate. The antimony composition of the Bi1 – xSbx layer is perfectly controlled and covers almost the whole TI window. For optimized growth conditions, the sample generates a semiconductor band structure at room temperature in the bulk and exhibits metallic surface states at 77 K.
_______________________________________________
Plasmonic-Induced Luminescence of MoSe2 Monolayers in a Scanning Tunneling Microscope
R. PECHOU, S. JIA, J. RIGOR, O. GUILLERMET, G. SEINE, J. LOU, N. LARGE, A. MLAYAH, R. CORATGER
ACS Photonics 2020, 7, 11, 3061
 We report on a scanning tunneling microscopy-induced luminescence in MoSe2 monolayers supported by uniform and nanopatterned gold substrates. Luminescence intensity mappings, recorded with a nanometric spatial resolution, and spectroscopy measurements were performed and analyzed in terms of photon emission processes taking place within the tip–surface gap region, which supports strongly localized optical and electronic states. We found that, excited by tunneling electrons, the light emission is due to the radiative recombination of free excitons confined within the MoSe2 monolayer. Light emission is observed at positive and negative bias voltages with very different emission rates. The results are interpreted in terms of charge carrier injection in the MoSe2 layer. Additionally, electrodynamic simulations also stress that resonance between the emitted radiation and the surface plasmons formed in the tip–surface gap region plays a critical role in the emission process. When the MoSe2 layer lays on a nanopatterned gold substrate, its luminescence intensity, induced by the tunneling electrons, is enhanced by nearly an order of magnitude. Such an effect is observed here for the first time in scanning tunneling microscopy experiments. The luminescence enhancement is attributed to the surface plasmon properties of the nanopatterned gold substrate, which spectrally match the excitonic transition of the MoSe2 layer. We show that this surface-enhanced scanning tunneling microscope-induced light emission (SESTM-LE) effect is very useful for investigating the photon emission from localized emitters (e.g., quantum wells, quantum dots, molecules) with a subnanometer spatial resolution.
We report on a scanning tunneling microscopy-induced luminescence in MoSe2 monolayers supported by uniform and nanopatterned gold substrates. Luminescence intensity mappings, recorded with a nanometric spatial resolution, and spectroscopy measurements were performed and analyzed in terms of photon emission processes taking place within the tip–surface gap region, which supports strongly localized optical and electronic states. We found that, excited by tunneling electrons, the light emission is due to the radiative recombination of free excitons confined within the MoSe2 monolayer. Light emission is observed at positive and negative bias voltages with very different emission rates. The results are interpreted in terms of charge carrier injection in the MoSe2 layer. Additionally, electrodynamic simulations also stress that resonance between the emitted radiation and the surface plasmons formed in the tip–surface gap region plays a critical role in the emission process. When the MoSe2 layer lays on a nanopatterned gold substrate, its luminescence intensity, induced by the tunneling electrons, is enhanced by nearly an order of magnitude. Such an effect is observed here for the first time in scanning tunneling microscopy experiments. The luminescence enhancement is attributed to the surface plasmon properties of the nanopatterned gold substrate, which spectrally match the excitonic transition of the MoSe2 layer. We show that this surface-enhanced scanning tunneling microscope-induced light emission (SESTM-LE) effect is very useful for investigating the photon emission from localized emitters (e.g., quantum wells, quantum dots, molecules) with a subnanometer spatial resolution.
_______________________________________________
Control of the deprotonation of Terephthalic Acid assemblies on Ag(111) studied by DFT calculations and low temperature scanning tunneling microscopy
J. HEINZ, C. DURAND, H. TANG, R. CORATGER
Phys. Chem. Chem. Phys 2020, 22, 3173
 This paper deals with the investigations of terephthalic acid (TPA) molecules deposited on a low reactive Ag(111) surface and studied using scanning tunneling microscopy (STM) at low temperature and DFT calculations. These investigations show that two deprotonation states energetically equivalent can be produced at the single molecule level. On self-assemblies, the mobility of H atoms at 77 K favours the motion of created defects in the layer. STM observations and DFT calculations show that the most stable structures are obtained when only one hydrogen atom is removed from an O–H…O bond and when these deprotonated molecules are located in adjacent TPA rows.
This paper deals with the investigations of terephthalic acid (TPA) molecules deposited on a low reactive Ag(111) surface and studied using scanning tunneling microscopy (STM) at low temperature and DFT calculations. These investigations show that two deprotonation states energetically equivalent can be produced at the single molecule level. On self-assemblies, the mobility of H atoms at 77 K favours the motion of created defects in the layer. STM observations and DFT calculations show that the most stable structures are obtained when only one hydrogen atom is removed from an O–H…O bond and when these deprotonated molecules are located in adjacent TPA rows.
_______________________________________________
8-Hydroxyquinoline (8Hq) complexes on Al(111): atomic scale structure, energetics and charge distribution
Yann Bulteau, Nathalie Tarrat, Nadine Pébère and Corinne Lacaze-Dufaure
 8-Hydroxyquinoline (8Hq) is known to efficiently inhibit the corrosion of aluminium by forming metal–organic layers (8Hq forms complexes with aluminium atoms). In the present work, the atomic scale structure and the energetics of 8-hydroxyquinoline complexes (Alq3) adsorbed on an aluminium surface are investigated by dispersion-corrected DFT computations. Two scenarios are considered: (i) an Alq3 complex, previously formed in vacuum, is deposited on a flat Al(111) surface or (ii) three deprotonated 8Hq molecules (q) directly adsorb on a defective Al(111) surface presenting Al adatoms (Al–Al(111)). For the Alq3 formation in vacuum, each addition of a q molecule on the Al atom stabilises the system, the oxidation state of the Al atom evolving from AlI in Alq to AlIII in Alq2 and Alq3. The subsequent deposition of Alq3 on Al(111) leads to a strong bonding between the q molecules of the complex and the Al(111) surface, with a significant electron transfer occurring from the surface to the complexes (0.73 to 1.57 e). The formation on the metal surface of Alq3 complexes via the adsorption of q molecules on an Al adatom leads to more stable structures than the ones obtained from direct adsorption of Alq3 on Al(111). For the most stable adsorption conformation, the three q molecules are bonded to the Al adatom but only two are bonded to the aluminium surface. In that case, the total electron transfer from the Al–Al(111) surface to the q molecules is 4.40 e and the electron transfer from the Al(111) surface to the Alq3-like species is 2.04 e. The structure, energetics and charge distribution data demonstrate an iono-covalent bonding between the q molecules and the Al atoms, in the complex as well as on the aluminium surface.
8-Hydroxyquinoline (8Hq) is known to efficiently inhibit the corrosion of aluminium by forming metal–organic layers (8Hq forms complexes with aluminium atoms). In the present work, the atomic scale structure and the energetics of 8-hydroxyquinoline complexes (Alq3) adsorbed on an aluminium surface are investigated by dispersion-corrected DFT computations. Two scenarios are considered: (i) an Alq3 complex, previously formed in vacuum, is deposited on a flat Al(111) surface or (ii) three deprotonated 8Hq molecules (q) directly adsorb on a defective Al(111) surface presenting Al adatoms (Al–Al(111)). For the Alq3 formation in vacuum, each addition of a q molecule on the Al atom stabilises the system, the oxidation state of the Al atom evolving from AlI in Alq to AlIII in Alq2 and Alq3. The subsequent deposition of Alq3 on Al(111) leads to a strong bonding between the q molecules of the complex and the Al(111) surface, with a significant electron transfer occurring from the surface to the complexes (0.73 to 1.57 e). The formation on the metal surface of Alq3 complexes via the adsorption of q molecules on an Al adatom leads to more stable structures than the ones obtained from direct adsorption of Alq3 on Al(111). For the most stable adsorption conformation, the three q molecules are bonded to the Al adatom but only two are bonded to the aluminium surface. In that case, the total electron transfer from the Al–Al(111) surface to the q molecules is 4.40 e and the electron transfer from the Al(111) surface to the Alq3-like species is 2.04 e. The structure, energetics and charge distribution data demonstrate an iono-covalent bonding between the q molecules and the Al atoms, in the complex as well as on the aluminium surface.
_______________________________________________
Performances of the Lamb Model to Describe the Vibrations of Gold Quantum-Sized Clusters
Quentin Martinet, Alice Berthelot, Adrien Girard, Baira Donoeva, Clothilde Comby-Zerbino, Elodie Romeo, Franck Bertorelle, Marte van der Linden, Nathalie Tarrat, Nicolas Combe and Jérémie Margueritat
J. Phys. Chem. C 2020, 124, 35, 19324
Lamb modes describe the vibrations of an object as a whole from the stellar scale to the nanometer one. Lamb description has been built from the linear elasticity theory and considers a homogeneous elastic sphere. Our work tries to determine the minimum scale where this description remains valid by studying the vibration of quantum-sized gold clusters (Au6, Au9, and Au25) stabilized by organic molecules. First, our work shows that experimental frequencies of small-functionalized gold clusters obtained by low-frequency Raman spectroscopy can be interpreted with density functional theory calculations. Moreover, the Lamb model broadly succeeds in predicting these Raman acoustic modes only if a correction considering the mass of the surrounding ligands is added. Ligands affect vibrational modes of the core by their mass but also by their covalent bond with the core. The unexpected consequence of this electronic stabilization by the ligands is the sustainability of the Lamb description for clusters as small as six atoms. Finally, the limit of the Lamb model can be reached out at low temperature where the vibration mode spectrum presents a substructuration that the Lamb description, developed for a homogeneous sphere, is unable to predict.
_______________________________________________
Methoxy radical adsorption on gold nanoparticles: a comparison with methanethiol and methylamine radicals
Xavier Fenouillet, Magali Benoit, Nathalie Tarrat
 The adsorption of a methoxy radical (OCH3) on the low-energy flat gold surfaces Au(111), Au(100) and Au(110) and on surface defects (adatoms) was compared with those of methanethiol (SCH3) and methylamine (NHCH3) radicals. Using dispersion-corrected DFT, we showed that the adsorption energy of OCH3 on gold is significantly lower than that of SCH3 but not very far from that of NHCH3. Whatever the molecule, we found that the adsorption energy is of the same order on Au(110) and Au(100), and smaller on Au(111) and that the charge transfer goes from the surface to the molecule. The charge transferred to SCH3 is very small, while that transferred to NHCH3 is slightly larger, but still three times smaller than in the case of OCH3. Concerning the competition between adsorption sites, we observed that undercoordinated atoms are not systematically more favoured than flat surfaces.
The adsorption of a methoxy radical (OCH3) on the low-energy flat gold surfaces Au(111), Au(100) and Au(110) and on surface defects (adatoms) was compared with those of methanethiol (SCH3) and methylamine (NHCH3) radicals. Using dispersion-corrected DFT, we showed that the adsorption energy of OCH3 on gold is significantly lower than that of SCH3 but not very far from that of NHCH3. Whatever the molecule, we found that the adsorption energy is of the same order on Au(110) and Au(100), and smaller on Au(111) and that the charge transfer goes from the surface to the molecule. The charge transferred to SCH3 is very small, while that transferred to NHCH3 is slightly larger, but still three times smaller than in the case of OCH3. Concerning the competition between adsorption sites, we observed that undercoordinated atoms are not systematically more favoured than flat surfaces.
_______________________________________________
Polymorphism in carbohydrate self-assembly at surfaces: STM imaging and theoretical modelling of trehalose on Cu(100)
Sabine Abb, Nathalie Tarrat, Juan Cortés, Bohdan Andriyevsky, Ludger Harnau, J. Christian Schön, Stephan Rauschenbach and Klaus Kern
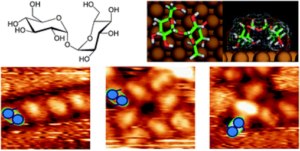 Saccharides, also commonly known as carbohydrates, are ubiquitous biomolecules, but little is known about their interaction with surfaces. Soft-landing electrospray ion beam deposition in conjunction with high-resolution imaging by scanning tunneling microscopy now provides access to the molecular details of the surface assembly of this important class of bio-molecules. Among carbohydrates, the disaccharide trehalose is outstanding as it enables strong anhydrobiotic effects in biosystems. This ability is closely related to the observed polymorphism. In this work, we explore the self-assembly of trehalose on the Cu(100) surface. Molecular imaging reveals the details of the assembly properties in this reduced symmetry environment. Already at room temperature, we observe a variety of self-assembled motifs, in contrast to other disaccharides like e.g. sucrose. Using a multistage modeling approach, we rationalize the conformation of trehalose on the copper surface as well as the intermolecular interactions and the self-assembly behavior.
Saccharides, also commonly known as carbohydrates, are ubiquitous biomolecules, but little is known about their interaction with surfaces. Soft-landing electrospray ion beam deposition in conjunction with high-resolution imaging by scanning tunneling microscopy now provides access to the molecular details of the surface assembly of this important class of bio-molecules. Among carbohydrates, the disaccharide trehalose is outstanding as it enables strong anhydrobiotic effects in biosystems. This ability is closely related to the observed polymorphism. In this work, we explore the self-assembly of trehalose on the Cu(100) surface. Molecular imaging reveals the details of the assembly properties in this reduced symmetry environment. Already at room temperature, we observe a variety of self-assembled motifs, in contrast to other disaccharides like e.g. sucrose. Using a multistage modeling approach, we rationalize the conformation of trehalose on the copper surface as well as the intermolecular interactions and the self-assembly behavior.
_______________________________________________
Carbohydrate self-assembly at surfaces: STM imaging of sucrose conformation and ordering on Cu(100)
Sabine Abb, Nathalie Tarrat, Juan Cortés, Bohdan Andriyevsky, Ludger Harnau, J. Christian Schön, Stephan Rauschenbach and Klaus Kern
Angewandte Chemie Int. Ed. 2019, 58, 8336
 Saccharides are ubiquitous biomolecules but little is known about their interaction with and assembly on surfaces. Combining preparative mass spectrometry with scanning tunneling microscopy, we have been able to address the conformation and self‐assembly of the disaccharide sucrose on the Cu(100) surface with subunit‐level imaging. By combining a multi‐stage modelling approach and experimental data, we can rationalize the conformation on the surface as well as the interactions between the sucrose molecules, yielding models of the observed self‐assembled patterns on the surface.
Saccharides are ubiquitous biomolecules but little is known about their interaction with and assembly on surfaces. Combining preparative mass spectrometry with scanning tunneling microscopy, we have been able to address the conformation and self‐assembly of the disaccharide sucrose on the Cu(100) surface with subunit‐level imaging. By combining a multi‐stage modelling approach and experimental data, we can rationalize the conformation on the surface as well as the interactions between the sucrose molecules, yielding models of the observed self‐assembled patterns on the surface.
_______________________________________________
Interaction between perylene-derivated molecules observed by low temperature scanning tunneling microscopy
L. VERNISSE, O. GUILLERMET, A. GOURDON, R. CORATGER
 Derivative perylene molecules deposited on Ag(111) and on NaCl(001) ultrathin layers have been investigated using low temperature STM and NC-AFM. When the metallic substrate is held at ambient temperature during evaporation, the molecules form characteristic trimers on the Ag(111) surface and interact through their polar groups. Close to the steps, the molecules form linear structures and seems to stand side by side. On the other hand, after deposition on a substrate cooled at liquid helium temperature, single molecules are observed both on metal and on NaCl. On the ultrathin insulator layers, the STM images present characteristic contrasts related to the molecular orbitals which favors the localization of aldehyde groups. In this case, the lateral molecular interactions may induce the formation of small assemblies in which the electronic levels are slightly shifted. A possible interpretation of this phenomenon is to take into account polar interactions and charge transfer between neighboring molecules.
Derivative perylene molecules deposited on Ag(111) and on NaCl(001) ultrathin layers have been investigated using low temperature STM and NC-AFM. When the metallic substrate is held at ambient temperature during evaporation, the molecules form characteristic trimers on the Ag(111) surface and interact through their polar groups. Close to the steps, the molecules form linear structures and seems to stand side by side. On the other hand, after deposition on a substrate cooled at liquid helium temperature, single molecules are observed both on metal and on NaCl. On the ultrathin insulator layers, the STM images present characteristic contrasts related to the molecular orbitals which favors the localization of aldehyde groups. In this case, the lateral molecular interactions may induce the formation of small assemblies in which the electronic levels are slightly shifted. A possible interpretation of this phenomenon is to take into account polar interactions and charge transfer between neighboring molecules.
_______________________________________________
On the role of intermolecular interactions in stabilizing AuNP@Ampicillin nano-antibiotics
Xavier Fenouillet, Magali Benoit and Nathalie Tarrat
 The fight against antibiotic resistance has become a major public health issue in recent years. In this context, nano-antibiotics composed of gold nanoparticles with antibiotics grafted on their surface often exhibit outstanding properties, sometimes even bypassing bacterial resistance mechanisms. Among these nano-antibiotics, gold nanoparticles/ampicillin hybrid systems (AuNPs@Ampicillin) are very effective. However, despite their very promising antibacterial properties, very little information concerning their atomic-scale structure is reported in the literature. In the present paper, the structure and energetics of AuNPs@Ampicillin nano-antibiotics have been investigated using first-principles numerical simulations through the study of the ampicillin adsorption on the three low Miller index facets Au(111), Au(100) and Au(110) of the AuNPs as a function of both the antibiotics coverage and its protonation state. Intermolecular interactions were found to be very stabilizing for coverages compatible with experimental data. An optimal coverage zone has been determined, in which the combination of a favorable gold surface-antibiotics interaction and of stabilizing intermolecular interactions can lead to an overall stabilization of the nano-antibiotics. As regards the mechanism of action of the nano-antibiotics, this study has confirmed that the active site of the free antibiotic is exposed to the solvent when the antibiotic is grafted onto the gold nanoparticle.
The fight against antibiotic resistance has become a major public health issue in recent years. In this context, nano-antibiotics composed of gold nanoparticles with antibiotics grafted on their surface often exhibit outstanding properties, sometimes even bypassing bacterial resistance mechanisms. Among these nano-antibiotics, gold nanoparticles/ampicillin hybrid systems (AuNPs@Ampicillin) are very effective. However, despite their very promising antibacterial properties, very little information concerning their atomic-scale structure is reported in the literature. In the present paper, the structure and energetics of AuNPs@Ampicillin nano-antibiotics have been investigated using first-principles numerical simulations through the study of the ampicillin adsorption on the three low Miller index facets Au(111), Au(100) and Au(110) of the AuNPs as a function of both the antibiotics coverage and its protonation state. Intermolecular interactions were found to be very stabilizing for coverages compatible with experimental data. An optimal coverage zone has been determined, in which the combination of a favorable gold surface-antibiotics interaction and of stabilizing intermolecular interactions can lead to an overall stabilization of the nano-antibiotics. As regards the mechanism of action of the nano-antibiotics, this study has confirmed that the active site of the free antibiotic is exposed to the solvent when the antibiotic is grafted onto the gold nanoparticle.
_______________________________________________
Adsorption of Terarylenes on Ag(111) and NaCl(001)/Ag(111) : A Scanning Tunneling Microscopy and Density Functional Theory Study
J. P. DELA CRUZ CALUPITAN, O. GALANGUAU, O. GUILLERMET, M. YENGUI, J. ETCHEVERRIA, X. BOUJU, T. NAKASHIMA, G. RAPENNE, R. CORATGER, T. KAWAI
J. Phys. Chem. C 2018, 122, 11, 5978
 Photoswitching materials are building blocks of next generation optoelectronic devices which may require molecule deposition on a solid substrate. However, molecule properties change upon adsorption due to surface-molecule interactions and symmetry considerations. Scanning tunneling microscopy (STM) and density functional theory (DFT) offer techniques to address interactions of functional molecules down to the single-molecular level on solid substrates. In this paper, we present a combined STM and DFT study of a tert-butyl functionalized terarylene molecule on Ag(111) and NaCl(001)/Ag(111) at ∼5 K. tert-Butyl groups aided in identifying three conformations of the compound upon adsorption on the surface. DFT calculations showed that two of these conformations refer to different adsorption geometries of the trans conformation in the gas phase. The other was assigned to the nonreactive cis conformation. For the first time, this conformation was isolated and imaged at the single-molecular level. Calculations further showed that aside from the electronic structure of the molecule, methyl groups sticking out of the surface are the origin of bright spots observed on the STM. On NaCl(001)/Ag(111), only the trans conformation was found and the mapping of occupied and unoccupied states of terarylenes was accomplished for the first time.
Photoswitching materials are building blocks of next generation optoelectronic devices which may require molecule deposition on a solid substrate. However, molecule properties change upon adsorption due to surface-molecule interactions and symmetry considerations. Scanning tunneling microscopy (STM) and density functional theory (DFT) offer techniques to address interactions of functional molecules down to the single-molecular level on solid substrates. In this paper, we present a combined STM and DFT study of a tert-butyl functionalized terarylene molecule on Ag(111) and NaCl(001)/Ag(111) at ∼5 K. tert-Butyl groups aided in identifying three conformations of the compound upon adsorption on the surface. DFT calculations showed that two of these conformations refer to different adsorption geometries of the trans conformation in the gas phase. The other was assigned to the nonreactive cis conformation. For the first time, this conformation was isolated and imaged at the single-molecular level. Calculations further showed that aside from the electronic structure of the molecule, methyl groups sticking out of the surface are the origin of bright spots observed on the STM. On NaCl(001)/Ag(111), only the trans conformation was found and the mapping of occupied and unoccupied states of terarylenes was accomplished for the first time.
The SINanO group is involved in various transverse themes within the laboratory.
Gap plasmon modes and plasmon-exciton coupling in a hybrid Au/MoSe2/Au tunneling junction Gap plasmon modes and plasmon-exciton coupling in a hybrid Au/MoSe2/Au tunneling junction
E. Alves, R. Péchou, R. Coratger and A. Malyah
The light-matter interaction between plasmonic nanocavity modes and excitons at the nanometer scale is here addressed in the scanning tunneling microscope configuration where an MoSe2 monolayer is located between the tip and the substrate. We investigate by optical excitation the electromagnetic modes of this hybrid Au/MoSe2/Au tunneling junction using numerical simulations where electron tunneling and the anisotropic character of the MoSe2 layer are taken into account. In particular, we pointed out gap plasmon modes and Fano-type plasmon-exciton coupling taking place at the MoSe2/Au substrate interface. The spectral properties and spatial localization of these modes are studied as a function of the tunneling parameters and incident polarization. The light-matter interaction between plasmonic nanocavity modes and excitons at the nanometer scale is here addressed in the scanning tunneling microscope configuration where an MoSe2 monolayer is located between the tip and the substrate. We investigate by optical excitation the electromagnetic modes of this hybrid Au/MoSe2/Au tunneling junction using numerical simulations where electron tunneling and the anisotropic character of the MoSe2 layer are taken into account. In particular, we pointed out gap plasmon modes and Fano-type plasmon-exciton coupling taking place at the MoSe2/Au substrate interface. The spectral properties and spatial localization of these modes are studied as a function of the tunneling parameters and incident polarization.
![]()
ANR BENDIS (2021-2025 LAPLACE, CEMES, LGC)
M. Benoit, P. Benzo, N. Tarrat
Interaction de cibles biologiques avec des couches minces diélectriques contenant des nanoparticules d’argent : vers des surfaces antimicrobiennes ajustables
https://anr.fr/Projet-ANR-21-CE09-0008
___________________________________________
ANR FENMAG (2020-2024 CEMES, LCC, LPCNO)
M. Respaud (coordinateur), P. Benzo, M. J. Casanove, P. Lecante
Nanoparticules de Fe16N2 pour la fabrication d’aimants permanents
https://anr.fr/Project-ANR-19-ASTR-0021
___________________________________________
Enjeux sociétaux INSA, projet DEFIANT (2021-2024 LAAS, CEMES, IMT, TBI)
N. Tarrat
An interdisciplinary approach to the design of effective nanoparticle-based antimicrobials.
___________________________________________
ANR Nanoslim (2018-2022 InPhyNi, CEMES, CP2M, GPM, LPhiA, ICGM, ICI-ECN)
P. Benzo
Mise en forme des nanoparticules pendant l’étirage des fibres optiques
https://anr.fr/Projet-ANR-17-CE08-0002
___________________________________________
ANR NucleFox

METSA (Réseau français de Microscopie Electronique à Transmission et Sonde Atomique) ___________________________________________
![]()
TALEM (Transpyrenean Associated Laboratory for Electron Microscopy), laboratoire européen associé entre le CEMES et l’INA de Saragosse (Espagne)
___________________________________________
![]()
GDR PULSE (Processus ULtimes en épitaxie de SEmiconducteurs) (2013-2022)
Contact scientifique A. Ponchet
___________________________________________

RFCT (Réseau Français de Chimie Théorique)
___________________________________________

Noeud « France Grand Sud-Ouest » du Centre Européen de Calcul Atomique et Moléculaire (CECAM-FR-GSO) – Direction M. Benoit
___________________________________________

GDR Or-Nano (L’or nanométrique : études fondamentales et applications pour la santé et l’environnement) (2017-2021) – Membre du bureau N. Tarrat
___________________________________________
Fédération TheMoSiA (Théories, Modélisations et Simulations Atomistiques) – Contact scientifique CEMES N. Tarrat
___________________________________________
GDR « Intelligence Artificielle en Science des Matériaux » – Directrice Adjointe M. Benoit

